

临床诊断实用指南《关于遗传性多囊肾疾病测序方案的选择》
多囊肾疾病简介:多囊肾是一种常见的遗传性疾病,全世界大约有400~600万患者,我国约有150万。多囊肾是引起肾功能不可逆下降的遗传性疾病,大部分患者最终需要肾脏替代治疗(透析)。多囊肾导致尿毒症的患者占血液透析患者总数的7%~10%。因为至今仍缺乏有效的防治手段,多囊肾一直是肾病领域的一个研究热点。
1
为什么会发生多囊肾?
多囊肾按照遗传方式可分为常染色体显性遗传性多囊肾(autosomal dominant polycystic kidney disease, ADPKD)和常染色体隐性遗传性多囊肾(autosomal recessive polycystic kidney disease, ARPKD)。其中,ARPKD是儿童期最常见的遗传性囊性肾病,该病发病率高,存活儿的发病率为1/20000[1]。ADPKD是慢性肾脏病最常见的遗传性疾病,在所有多囊肾病例中,其致病基因PKD1 突变(占71%-85%)比PKD2 突变(占大约15%)更常见。然而,还有大约6.8%的多囊肾患者并未发现有这两种基因突变。
一般常染色体显性遗传性多囊肾类型更常见,通常患者数十年没有任何症状和迹象,发病时多集中在30~40岁之间。因此,依照发病年龄,这种类型多囊肾又常被称为“成人型多囊肾”。
对于常染色体显性遗传性多囊肾疾病,只要父母双方中有一方具有致病的异常基因,孩子就会有50%的几率患上同样的疾病(如下图)。这样的情况,占多囊肾发病的90% ,其特点为具有家族聚集性,男女均可发病,两性受累机会相等,连续几代均可出现患者。成人型多囊肾是单基因病,PKD1 基因突变所致为Ⅰ型,PKD2 基因突变所致为Ⅱ型。Ⅰ型最为多见,占85%-90%。PKD1 基因突变的患者通常比PKD2 基因突变病情进展要快,但个体差异较大。

图片来源:google.com
常染色体隐性遗传性多囊肾比较少见,通常患儿在很小的时候就夭折了。另外,还可能存在其他未知的致病基因引起常染色体显性遗传性多囊肾。
2
多囊肾临床诊断
通常,结合家族史和常规超声检查(如下图)、CT等影像学检查结果可以确诊。
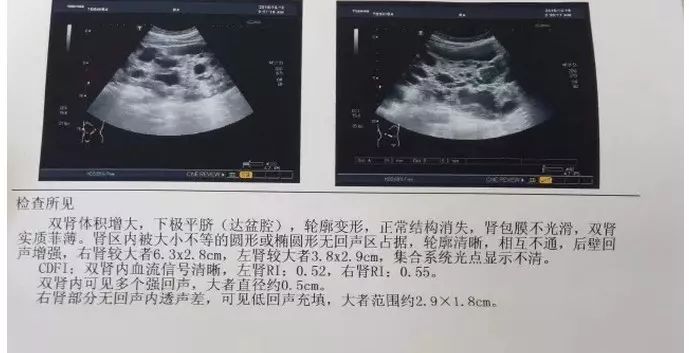
图片来源:google.com
随着技术的发展,目前通过基因测序,可以诊断携带多囊肾基因的患者,借助基因检测可以早期辅助临床确诊,对于有多囊肾家族史的家庭成员筛查,有利于早期发现多囊肾。如果多囊肾患者备孕,需要提前做好相关遗传咨询。
3
多囊肾基因检测方案
如若临床影像学检测怀疑多囊肾,但在分型不是很明确,对于出报告周期希望不要有延迟风险的情况下,建议首选做多囊肾基因特殊检测:
(1)PKD1 特殊检测:Long-PCR+一代测序
基因:PKD1(~85%,更常见)成人型(AD遗传)
PKD2 一代测序
基因:PKD2(~15%)成人型(AD遗传)
(2)儿童型(AR遗传)
多囊肾合并肝病
基因:PKHD1
既然多囊肾相关目标基因如此明确,那么为什么要选特殊检测呢?
原因在这3个目标基因里面,PKD1 基因存在假基因(假基因指与正常基因相似,但丧失正常功能的DNA序列,往往存在于真核生物的多基因家族中,是基因组中与编码基因序列非常相似的非功能性基因组 DNA 拷贝,一般情况都不被转录,且没有明确生理意义)。单纯的二代高通量基因测序不能区分基因变异来源真基因或假基因的问题。
假基因现象:
(1)PKD1 存在假基因现象:有6个同源区段,同源性高达97.7%(处于1-33号外显子)全外测序区分不了真假基因的问题。
(2)已知PKD1 的突变没有热点突变,广泛分布(见下图)
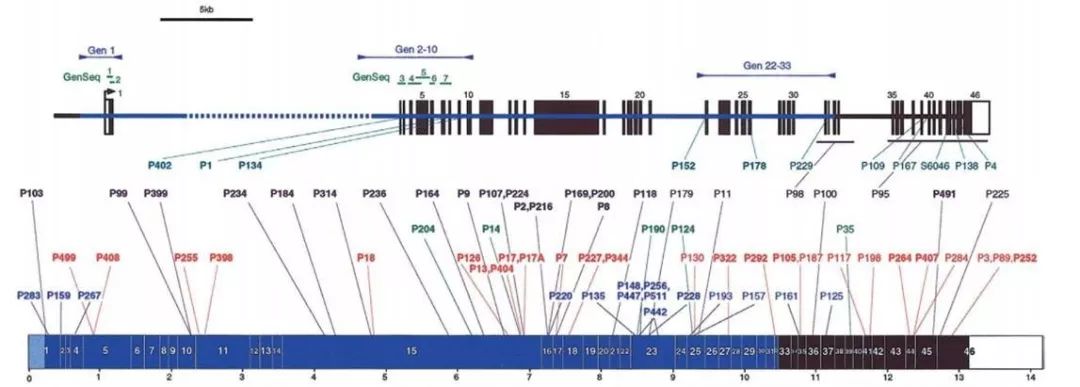
图片来源:AM J HUM GENET
由于假基因的存在,对全外测序检测准确找出真实的突变致病位点带来了挑战。全外检测出的位点变异很可能存在假阳性,不具有临床参考意义。
小结
多囊肾疾病检测方案选择如下图:
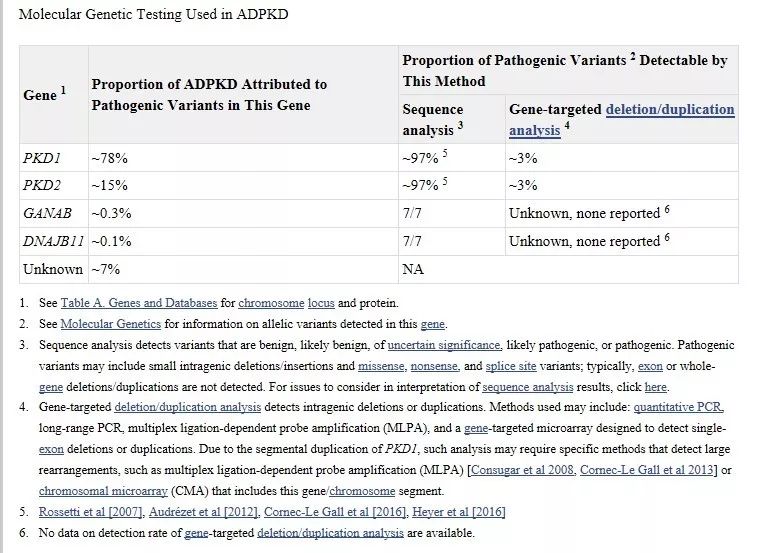
图片来源:geneReviews
若临床明确怀疑患者为多囊肾疾病,尽量选择多囊肾特殊检测(PKD1 特殊检测+PKD2 一代补测)[2]。
最全面检测方案:多囊肾特殊检测(PKD1 特殊检测+PKD2 一代补测)+全外显子
多囊肾基因检测意义:
决定各个患者病情进展速度的主要因素为致病的基因突变类型。PKD2 型患者比PKD1 型患者进展更缓慢。一项回顾性研究证明了这一点,该研究[3]分别纳入了333例PKD1 型患者和291例PKD2 型患者,与398例匹配对照者进行了比较。PKD1 型和PKD2 型患者发展至终末期肾衰竭的中位年龄分别为54岁和74岁。
2%-3%的患者同时缺失PKD1 基因和复合型结节性硬化症TSC2 基因,导致一种更严重的囊性肾脏病变,被称作相邻基因综合征。
其他遗传因素,如基因内突变的位点和类型,也可能会影响疾病的严重程度。与PKD1 的3'位点发生突变的患者相比,5'位点发生突变的患者发展至ESRD(end stage renal disease,终末期肾脏病)的年龄更小,并且肾脏外表现更严重。此外,PKD1 截短突变比错义突变导致的疾病更严重。目前还没有观察到PKD2 患者的疾病严重程度与基因突变位点有明确关联,但有一些特定的突变类型可能与病情较轻相关。
预测基因型 — 基因检测可用于识别多囊肾的致病性基因突变,但并不常规进行,除非必须做出确切诊断,如筛选家庭成员以寻找可能的肾脏供体时。
在有已知PKD 和基因型未知的多囊肾家族史的患者中,仔细关注家族史或可预测突变。
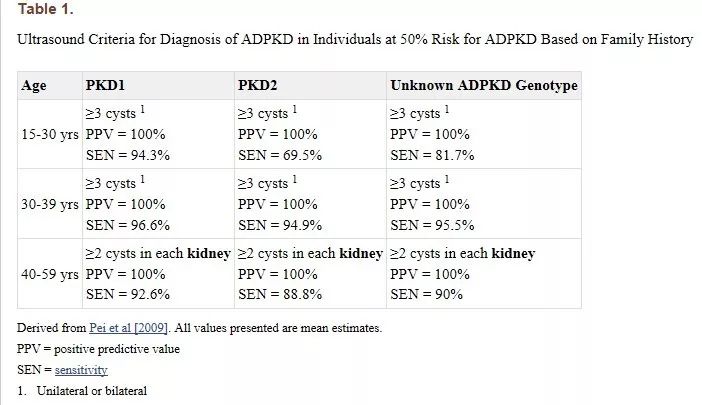
图片来源:geneReviews
参考文献:
【1】Parfrey, Patrick S . Autosomal-recessive polycystic kidney disease[J]. Kidney International, 2005, 67(4):1638-1648..
【2】Borràs, Daniel M, Vossen R , Liem M , et al. Detecting , PKD1 , variants in polycystic kidney disease patients by single-molecule long-read sequencing[J]. Human Mutation, 2017.
【3】Hateboer N , Dijk M A V , Bogdanova N , et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2[J]. Lancet (North American Edition), 1999, 353(9147):0-107.