

AJHG | Sin3/HDAC复合物成员SIN3B单倍体不足导致智力残疾综合征及自闭症

SIN3/HDAC复合物可通过与DNA 结合蛋白的相互作用来结合靶基因的启动子,也可直接结合转录因子,引起的组蛋白去乙酰化使得转录因子无法与 DNA 特异性结合,从而抑制转录。在哺乳动物中,编码SIN3复合物的基因有两个,分别为SIN3A和SIN3B,人类SIN3A突变导致伴有颅面缺陷的智力残疾综合征,而SIN3B突变所导致的疾病则鲜有报道。
2021年5月6日,法国南特大学医学遗传学中心与美国杜克大学人类疾病建模中心在The American Journal of Human Genetics 杂志上联合发表了名为“Haploinsufficiency of the Sin3/HDAC corepressor complex member SIN3B causes a syndromic intellectual disability/autism spectrum disorder” 的研究成果。该研究通过染色体微阵列或外显子组测序并结合国际数据共享工作,确定了9例具有杂合 SIN3B 缺失或单核苷酸变异的患者。本研究通过遗传学、表观遗传学及斑马鱼模型数据验证了SIN3B 致病突变导致神经发育综合征,其表现为生长缺陷、面部畸形、智力障碍、发育迟缓并伴有不同程度的自闭症谱系障碍。作者进一步指出表观遗传调节因子,特别是组蛋白乙酰化调控基因的突变,与多种神经发育障碍相关,说明了组蛋白翻译后修饰的严格调控在神经发育过程中的重要性。

该研究通过对9例携带罕见SIN3B变异的患者(7例新发,2例由于无法获得亲本样本而无法确定变异来源)的临床和脑部MRI数据的联合分析,报告了与SIN3B单倍剂量不足相关的表型。其中 1、2、9号患者表现出多动障碍,3、6号患者脑部成像显示胼胝体发育缺陷,4号患者表现为轻度扁桃体异位,7号患者从3岁开始出现全身强直阵挛发作,2、3、5号患者出现心室间隔缺损,2、3、4号患者伴随有唇腭裂的表型。
研究人员通过染色体微阵列分析发现有5例患者在19p13.11区域上存在拷贝数变异,以往的研究显示,19p13.11区域上的拷贝数变异涉及包括神经发育障碍在内的多系统疾病,目前已有两例拷贝数变异的案例报道,与本研究中的5 例患者相比较,产生了一个重叠区域,该区域包含NWD1、SIN3B、F2RL3、CPAMD8 四个基因(如图1A所示)。NWD1、F2RL3 基因未被报道过与人类遗传疾病相关;CPAMD8 双等位基因突变导致眼前节发育不良,但未有神经发育症状;而在智力残疾患者中发现了SIN3B基因的可能有害变异。因此,本研究针对SIN3B基因突变对神经发育的影响展开研究。
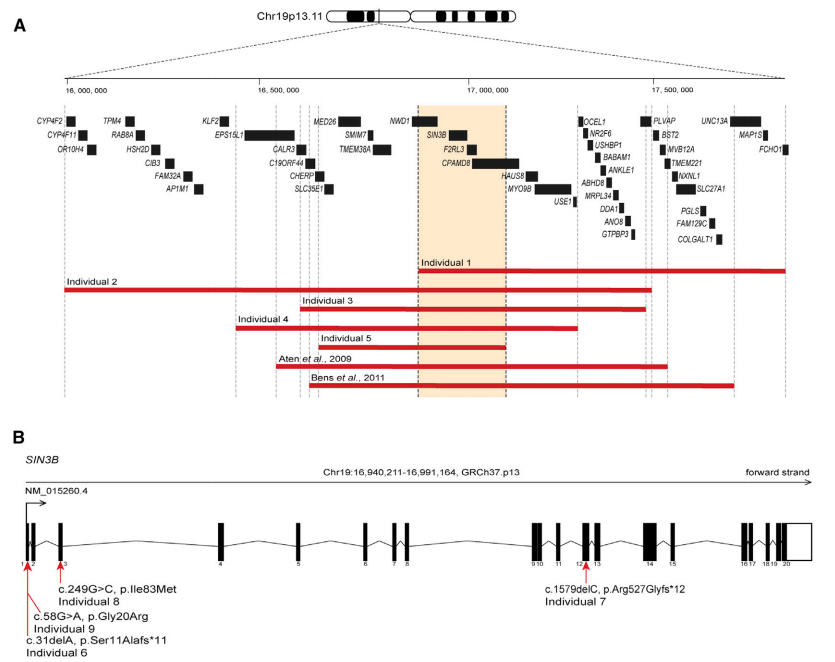
该研究中报告的6号患者在SIN3B的第一个外显子处存在移码突变(c.31delA [p.Ser11Alafs*11]),预测该突变会诱发无义介导的mRNA降解,导致SIN3B单倍剂量不足,但无法获得患者细胞系进行验证实验。
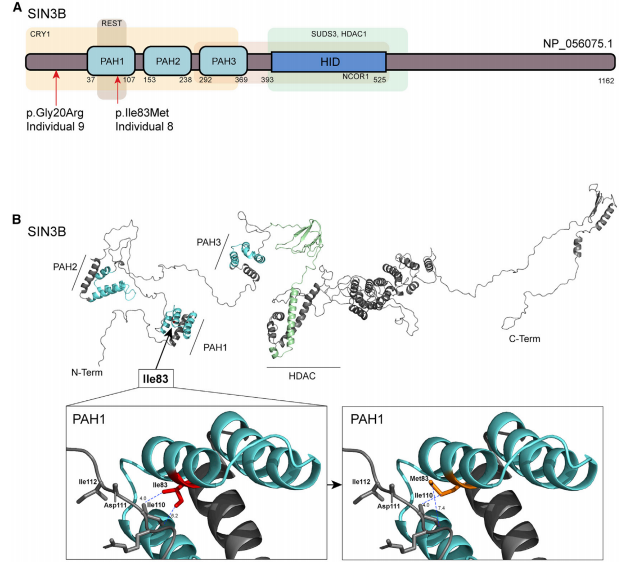
8号患者的外显子测序(ES)发现一个新发突变(c.249C>G [p.Ile83Met]),蛋白结构预测为有害突变,这一突变影响SIN3B的两亲性螺旋结构域(PAH)中的保守残基,该结构域负责与SIN3/HDAC复合物成员间的相互作用(如图2所示)。
9号患者存在杂合突变(c.58G>A [p.Gly20Arg]),这一突变在gnomAD中未被收录,但蛋白结构预测为有害突变,在排除该患者携带的其他可能突变致病的情况下,作者认为该患者的SIN3B突变可能与其神经发育表型相关。
利用转基因斑马鱼进行基因编辑
上述斑马鱼实验数据都证实了SIN3B单倍体不足对生长发育产生了有害影响,验证了人类SIN3B 基因突变导致的临床表型。
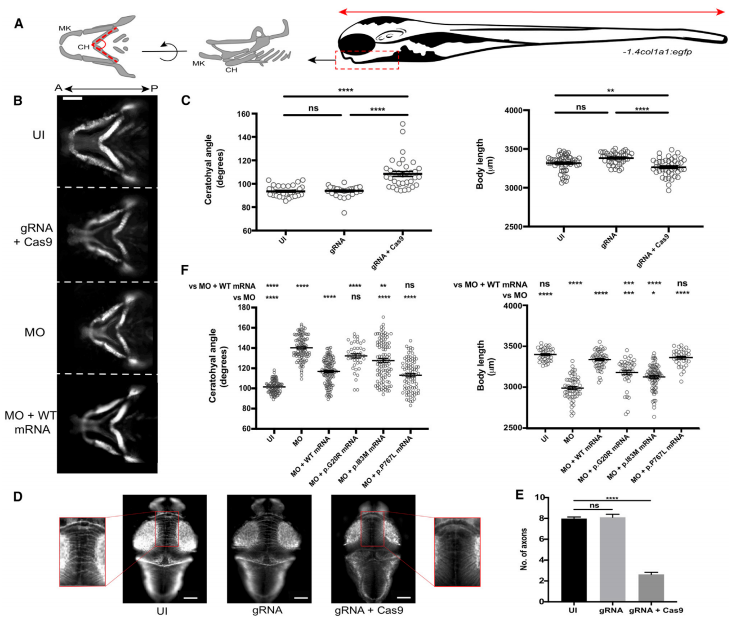
图3 sin3b缺失引起颅面缺陷、
体长减少和神经发育异常
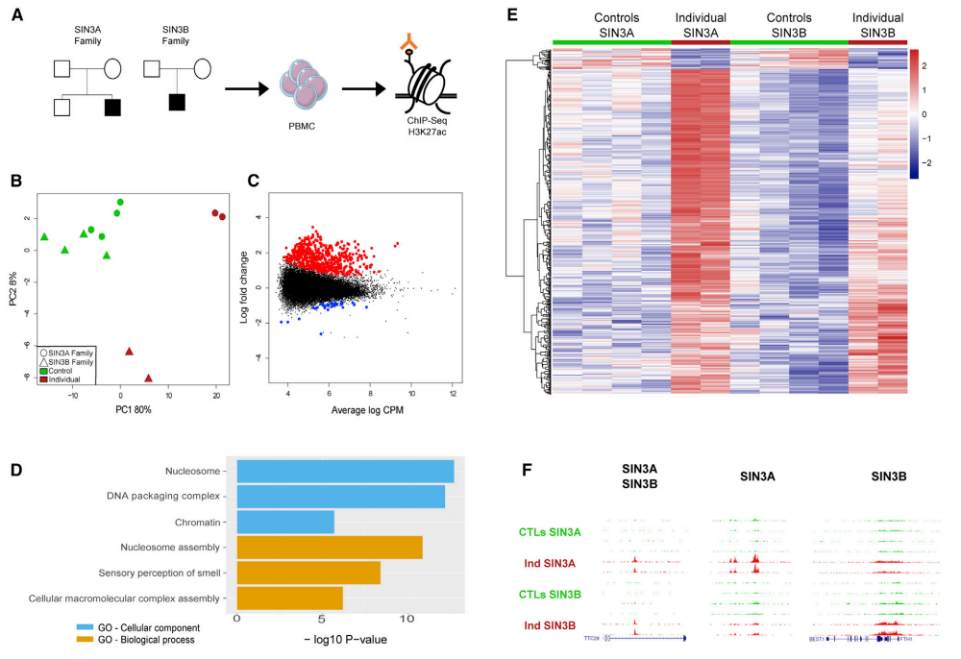
SIN3B突变对基因调控的影响
参考文献
https://www.cell.com/ajhg/fulltext/S0002-9297(21)00101-4
赛福基因现已开展“功能验证”服务,欢迎添加客服微信了解详情。
![]() 微信扫描/长按识别二维码:
微信扫描/长按识别二维码:
