

赛福基因公开课《Rett综合征》


各位老师好,我是范珊珊。今天很荣幸和各位老师一起探讨一下与Rett Syndrome相关的一些遗传学知识。

Rett综合征最初是由奥地利的Andres Rett博士于1966首次报道,当时发现有22名儿童,均是女孩,表现出部分获得性技能丧失,比如手部技能障碍和交流障碍,这种严重的神经系统发育异常性疾病,就以首次发现者的医生名字命名,称为Rett 综合征(RS,RTT)。20年后,随一份来自瑞典、葡萄牙和法国的35名受影响女孩的报道后广为人知。
Rett综合征,主要累及女性,被认为是继Down综合征之后,影响女性智力发育的第二大重要原因。发病率为 1/10000~1/15000。

女性RTT患者通常有正常的出生史和发育史。
女性患者的RTT病情进展通常会表现4个阶段:
阶段I,早期起病期,一般出生后6~18个月起病。这一阶段的发育迟缓可能由于症状较模糊而被家长所忽视。但回顾性数据分析表明,这些儿童中的大多数在婴儿早期有细微的行为差异。他们被描述为非常平静,呼吸、哭声微弱这一阶段通常持续几个月,但有些可持续一年以上。
阶段II,病情迅速进展期,通常在1岁~4岁之间。可表现为语言功能降低或者丧失,手的技能降低,且部分患儿存在单一模式手的刻板动作;坐和走的功能受限,自闭症的行为,呼吸不规律,头部发育落后等。
阶段III,病情稳定期,通常在2岁~10岁之间,表现为运动障碍,癫痫发作;行为有所改善(烦躁,哭缓解),警觉性,注意力,和沟通技巧有所改善。一些严重的女性患者可能会在该阶段病逝。
阶段IV期,或晚期运动恶化阶段,行动不便、肌肉无力、强直、痉挛,肌张力障碍。总的来说,在该阶段不会观察到认知、沟通、或手部技能下降表现,但会常见进食障碍和体重增加困难。

RTT患儿有许多临床表型,且与其他综合征有重叠的表型,给临床诊断带来了困难。诊断RTT主要依靠临床表现。2001 年的诊断标准中: 含有 8 条必需标准、5 条排除标准和 8 条支持标准,以上标准都没有准确描述必要条件之一———生后头围增长减速。因为不是每个典型的RTT患儿都有生后头围增长减速,但为了不漏诊,2010 年的国际RTT临床研究协会提出新的修订版诊断标准,诊断标准将生后头围增长减速放置于所有诊断指标之前,强调有此表现就应该疑诊RTT。2010 年的诊断标准中对支持标准不作要求,对诊断典型的RTT更简单、确切。

在RTT患者当中,有一个超级罪犯基因MECP2,在已诊断明确的的典型RTT女性患者当中,携带有MECP2基因点突变/小片段插入缺失突变的患者比例为80%左右,8%是由于该基因的部分或整个基因缺失,剩余部分可能是由于其他变异,比如CDKL5或FOXG1导致。
非典型RTT患者该比例携带有MECP2基因点突变/小片段插入缺失突变的患者比例为40%左右,3%是由于该基因的部分或整个基因缺失导致。
常见的突变类型包括:错义突变、无义突变、移码突变、插入/缺失、以及部分或整个基因的缺失。

MECP2基因编码X连锁的甲基化CpG结合蛋白-2,是一种高丰度的染色质结合蛋白。MECP2不仅可以通过调节DNA甲基化途径影响甲基化过程,还可以作为转录抑制因子调节基因转录,从而维持修饰神经元成熟。因此,如果MECP2基因产生突变导致其编码的蛋白功能异常,可能对神经系统的生长和发育造成损害。
该蛋白共有486个氨基酸,包含2个主要功能区:MBD和TRD。
MBD区域,含有85个氨基酸,为蛋白质染色质定位所需 ,该突变可能改变蛋白的二级结构,导致MECP2蛋白与5-甲基胞嘧啶的结合能力下降。
TRD功能区,由104个氨基酸组成,它与组蛋白脱乙酰化酶和转录因子SIN3A相互作发挥转录抑制作用。
MECP2中已知的致病变异约有80-85%的变异位于MBD、TRD和C端末区。
RettBASE是由澳大利亚的Westmead儿童医院建立,网站中记录了所有的和RTT疾病相关的所有基因的突变信息。
据该网站记录,目前已知的与RTT疾病相关的MECP2基因变异共有896项。

为了探讨中国RTT患者与MECP2这个基因的关系,2007年,国内的一篇报道对121名RTT患者进行了MECP2基因的测序分析,整体的实验策略是先对121名患者的MECP2基因的2-4号外显子区进行测序,如果没有发现致病突变,则对1号外显子进行测序分析。若仍为阴性结果,则使用MLPA的方法检测MECP2基因的大片段缺失。
分析结果显示:
在121名RTT患者当中,MECP2基因的总体检出率是84.3%,其中典型RTT的MECP2的检出率是87.9%,非典型RTT的检出率是57.1%。
在检出的突变位点当中,T158M突变型最为常见,所占比例是15.7%,随后是R168X (11.8%)。
突变类型当中,错义突变(43.1%)、无义突变(26.5%)、移码突变(17.6%)。
1号外显子中检出突变所占比例稀少,仅有0.9%。
大片段缺失检出率是10.5%,所有的缺失突变均出现在exon3、exon4或2者都有,表型严重程度和缺失的片段大小无相关性。

不知道大家有没有注意到一个问题,RTT患者为何累及的多为女性?

这还是要从RTT患者当中的超级罪犯基因MECP2说起:
MECP2基因位于X染色体Xq28区域,其对应的遗传方式是属于X连锁显性遗传(XLD)。
以前我们介绍过染色体的概念,每个人是由23对染色体组成,其中决定男女性别的主要是第23对性染色体,女性是XX,男性是XY。单基因遗传病的遗传方式,按照致病基因是位于常染色体还是性染色体,可以分成2大类:常染色体遗传疾病,和性染色体连锁遗传病。
其中性连锁遗传病就是由于性染色体上的基因所决定的性状,在群体分布上存在明显的性别差异。
如果决定一种遗传病的致病基因位于X染色体上,并随X染色体在上下代之间传递,我们就称之为X连锁遗传病。
X连锁显性遗传病的定义的就是等位基因之一突变,杂合状态下即可发病。
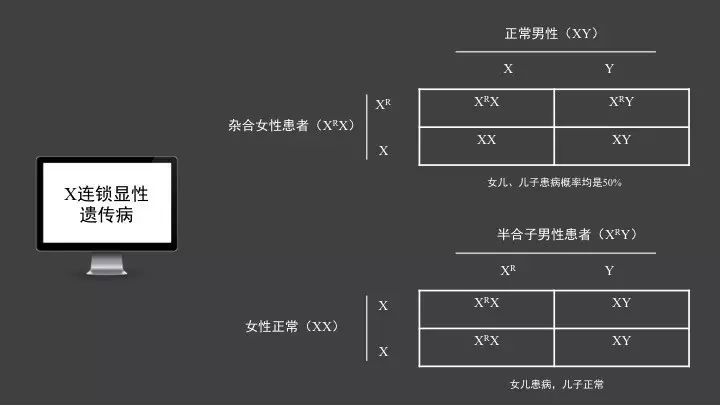
X连锁显性遗传病的女性杂合子患者和正常男性婚配,类似于常染色体显性遗传,其将致病基因遗传给儿子和女儿的概率均是50%。
男性只有一条X染色体,其X染色体的等位基因不是成对存在的,在Y染色体上缺少相对应的等位基因,称为半合子,其X染色体上的基因都可表现出相应的症状或疾病。X连锁显性遗传病中的男性患者,其致病基因一定会传给女儿,而不会传给儿子,故女儿都将是患者,儿子全部正常。

那么对于RTT疾病来说,为何报道的多为女性患者呢?
原因一是对于X连锁显性遗传病来说,女性有2条X染色体,其中任何一条X染色体上存在等位基因,就会发病,而男性只有一条X染色体,所以在生育率正常的情况下,家系中女性患者的数目要比男性患者多约一倍。
原因二是男性RTT患者,表型严重,多于出生后即出现严重的新生儿脑病,并于短期内死亡。那么为何X连锁显性遗传疾病中男性患者的病情会比女性严重呢?主要包括以下3点原因:
由于群体中致病基因频率很低,女性纯合子的概率很小,临床多见的女性一般都是患病杂合子;
女性杂合子患者由于还存在一个正常的等位基因,在不完全显性的情况下,病情比男性轻,且差异较大;
女性患者由于X染色体的随机失活,当带有致病基因的X染色体失活时病情较轻,且常有变化。

在罕见的幸存的男性,最常见的临床表现是严重的新生儿发病脑病与畸形,异常音、不自主运动、严重的癫痫发作,呼吸异常。
严重的新生儿脑病表型在女性RTT患者中很少见。
但是也有特例, Clayton-Smith等的一篇报道显示,男性RTT患者中,由于存在体细胞嵌合现象,导致即使携带了MECP2基因突变,也并没有引起严重的疾病表型。
体细胞嵌合现象也可以解释一些其他的X染色体显性遗传疾病出现在男性患者当中,但是没有引起致死的现象。

典型女性RTT或非典型RTT患者,即使均携带MECP2的杂合突变,其临床表型严重程度也各不相同,有些患者表型很轻,甚至不会出现,而有些则相当严重。
原因就是女性中存在的X染色体随机失活现象。
女性的两条X染色体在胚胎发育早期就有一条随机失活,称为X染色体失活或Lyon化;
对于女性为杂合子突变的X连锁显性遗传病来说,女性体内部分体细胞中带有致病基因突变的X染色体失活,另一部分未携带致病基因突变的X染色体失活。而男性患者中不存在这种机制,若为半合子突变,在不考虑体细胞嵌合的情况下,全身体细胞均是突变型,因此病情会很重。所以在X连锁显性遗传病中,女性杂合子患者的病症往往比男性患者的较轻。
但是由于X染色体失活是随机发生的,不同的女性RTT患者中X染色体失活的比例不同,所以在临床疾病表现程度上,会有轻重的区别。

那么对于检出MECP2基因突变的RTT患者,我们应如何进行遗传咨询呢?

对于女性患者双亲来说:
RTT临床99%为散发病例,1%有明确家系,散发病例绝大部分为新生MECP2基因突变,也可能遗传具有致病突变的生殖细胞嵌合体父亲或母亲,或遗传自由于X染色体随机失活而无或仅有轻度表现的携带者母亲,由于嵌合体父或母亲及携带者母亲无或仅有轻度临床表现,致使部分病例为临床散发。
如果已知女性由于X染色体随机失活而无或仅有轻度表现的携带者母亲,其后代携带MECP2致病突变的概率是50%。
男性患者的双亲:男性患者的父亲不会是携带者,母亲可能是由于X染色体随机失活而无或仅有轻度表现的携带者。
所以对于先证者基因型明确的家庭来说,其父母双亲最好进行一下分子遗传学的检测。

若先证者的双亲拟再生育,其生育健康小孩的概率也必须建立在先证者致病基因突变已明确,父母对应的基因位点也明确的基础上进行。
若检出先证者母亲是携带者,携带相同的MECP2基因突变,那么其将致病基因遗传给后代的概率是50%。
若双亲中均未携带相同的MECP2基因突变,那么可能出现以下2种情况:
一是先证者为denovo突变,那么双亲再生育患病后代概率将会非常低;
二是双亲中存在生殖细胞嵌合现象。

所以对于这种情况下,应该也是临床中遇见的比较多的情况,对于RTT患者的双亲来说,如果拟再生育,最好还是做一下产前检测。
2005年的一篇报道,显示已生育一携带MECP2突变的患病女儿,但双亲未携带的情况下,其再生育出与先证者女孩携有相同突变型的比例是1/9。

那么如果RTT患者想孕育后代,其生健康宝宝的概率又是如何呢?
只要是携带MECP2相关变异的RTT患者,将致病基因遗传给后代的概率均是50%,即使典型RTT患者不能再生育。
生育的携带者女孩,即使考虑到X染色体失活现象,患RTT疾病的概率仍是高风险。
生育的携带者男孩,很可能就会出现严重的新生儿脑病,即使存活1年,也会患有严重的智力障碍。

MECP2基因突变关联疾病除RTT之外还包括:X连锁隐性遗传的新生儿脑病、X连锁精神发育迟滞,以及遗传模式较为复杂的自闭症相关。
与MECP2相关的X连锁精神发育迟滞,女性患者的临床表型可能表现从轻度到重度智力残疾与躁狂抑郁精神病。受影响的男性通常有严重的智力障碍。
RTT患者,尤其是患者的临床表型中不存在小头畸形,抽搐或脊柱侧后凸畸形的患者,临床上可能被诊断为自闭症,但是MECP2基因并不能算是引起自闭症的主要基因。
如果男性患儿中出现了严重的新生儿脑病,或是重症肌无力,或存在X连锁智力障碍的家族史,则应该考虑MECP2基因突变相关的疾病。
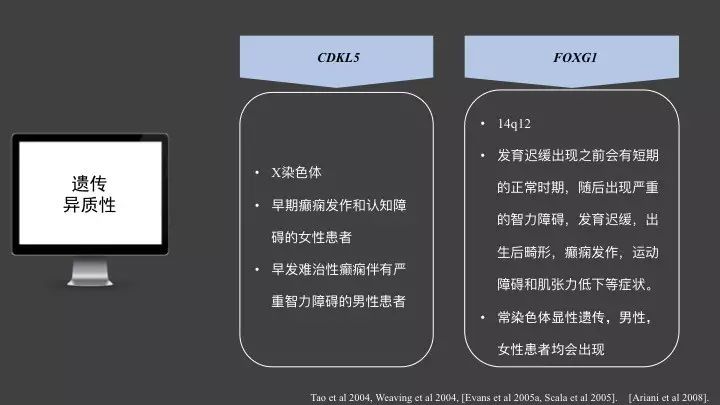
女性典型RTT患者当中,如果未检测到MECP2基因突变,在出生后6个月内出现癫痫发作,则可能携带有CDKL5基因突变。CDKL5最初是在伴有早期癫痫发作症状的RTT患者当中发现,在RTT-like患者当中,所占比例不是很高。现在普遍认为,CDKL5基因突变多与伴有早期癫痫发作和认知障碍的女性患者,以及早发难治性癫痫伴有严重智力障碍的男性患者中相关。
现已报道的与RTT疾病相关的CDKL5基因变异,据Rettbase数据库记载,也已有255项。
FOXG1,14q12,主要是在在发育迟缓出现之前会有短期的正常时期,随后出现严重的智力障碍,发育迟缓,出生后畸形,癫痫发作,运动障碍和肌张力低下等症状。这个基因呢是位于常染色体中,属于常染色体显性遗传,其对应的遗传模式和MECP2基因不同,男性,女性患者均会出现。所以对于存在这种基因突变的RTT患者,其对应的RTT表型在男性、女性中的分布以及遗传咨询均和以上所说的不同。这也体现了单基因遗传病的复杂情况。

现在已知的与RTT疾病关联的基因共有3个,分别是MECP2、CDKL5和FOXG1,但是临床中仍然有一部分RTT患者没有在这3个基因中发现致病突变,那么会不会存在新的RTT致病基因,我们还没有发现的可能呢?最新的一篇报道给了我们新的提示:
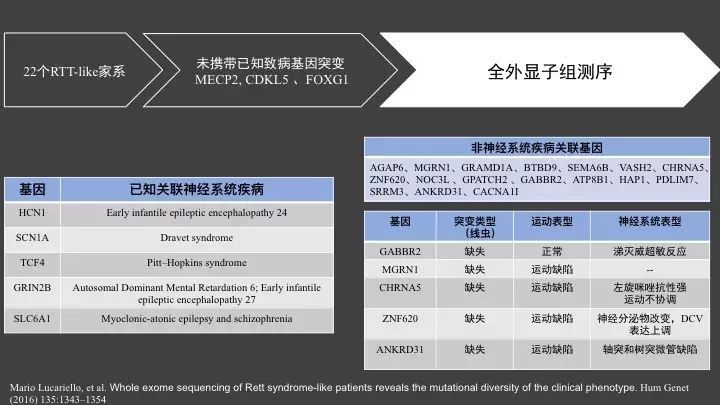
这是2016年发表在Hum Genet(5.138)杂志上的一篇文章,这篇文章对未检测到已知MECP2, CDKL5 、FOXG1致病基因突变,但临床表型存在刻板的手部动作,头小畸形,和12个月的年龄后发病的22个临床中怀疑是RTT疾病的家系进行了全外显子组测序。
共在14个家系中发现了致病基因突变,其中5个家系患者携带已知神经系统关联的致病基因,包括与早期婴儿癫痫脑病相关的HCN1基因,Dravet综合征相关的SCN1A基因等;
其他17个携带有以前未证明和神经系统疾病关联的致病基因突变,例如锚蛋白(Ankyrin)(一类富含锚蛋白重复序列功能域的蛋白);CHRNA5(神经元的乙酰胆碱受体α5亚单位)。
为了证明这些非神经系统关联疾病基因,表型和基因型的相关性,从这些基因中挑选了6个存在缺失突变的基因在线虫中进行了功能验证,由于RTT疾病多伴有运动障碍,实验中观察了线虫的运动和神经系统表型,结果发现,这些基因多可以引起线虫的运动功能缺陷。
所以WES仍是分析这些比较特殊病例的一个强有力的分析工具,而新的疾病关联基因的发现和证实,确实也是需要一个严谨而漫长的过程。

总之,对于RTT疾病来说,MECP2基因仍是我们仍需重点关注的超级罪犯基因,在对于RTT患者选用的检测方式中,如果从患者的经济利益考虑,我们最优推荐,针对MECP2基因进行2,3,4的外显子测序检测点突变和小插入缺失,再加上使用MLPA的检测方式进行大片段的检测。
但是这种情况下,若存在CDKL5 、FOXG1基因突变的RTT患者,就会漏检。
所以如果患者的经济条件允许,又比较着急的情况下,我们还是会推荐全外显子组测序+MLPA的组合检测方式,这样,RTT患者在分子诊断层面漏检的几率将会非常低。而且现在全外的测序成本越来越低,若临床中怀疑该疾病,但是没有确诊,患者的经济条件又允许的情况下,WES+MLPA将会是一个性价比最优的组合。

以上就是RTT疾病中的遗传学特征,及相应的遗传咨询和检测方式选择。感谢各位老师在百忙之中抽出时间收听我们这次课程。下面我们进入提问环节,各位老师有任何相关问题,都欢迎您在群内提出,我们共同进行交流。
1.如果针对MECP2基因进行一代测序+MLPA,这种方式的检出率大概是多少?
答:针对MECP2基因进行2,3,4的外显子测序检测点突变和小插入缺失,再加上使用MLPA的检测方式进行大片段的检测。那么根据已有的统计显示,该种检测方式的检出率约是:(~)80%+(~)10%。
2.现在RTT疾病中基因型和表型之间有对应关系吗?
答:目前已有的研究其实有挺多,但是一直没有一个相符的结果。有报道说,即使携带相同的基因突变,可能由于X染色体失活的机制,在表型上也是会有很大的差别。但是又有报道说,携带错义突变的患者表型会比无义突变的患者表型轻。

3.使用全外显发现新基因的那篇文章中,在RS疾病中发现的与神经系统不相关的那些基因都是和什么疾病相关的?
答:BTBD9—不安腿综合征,ATP8B1--家族性肝内胆汁淤积症。其他的好多基因,在OMIM数据库中还没有与已知疾病的报道,但是好多基因都与神经元生物学和功能相关,如神经元的乙酰胆碱受体亚单位CHRNA5,神经元电压门控性钙通道CACNA1等。