

深度内含子变异的SCN1A小鼠模型揭示该基因毒外显子异常调控在癫痫性脑病中的作用机制

Dravet综合征(DS)是一种发育性癫痫性脑病(DEE),其特点难治性癫痫发作、发育迟缓、语言障碍、共济失调、低张力、睡眠障碍和其他健康问题。编码电压门控钠离子通道(Nav1.1)SCN1A的功能缺失突变是引起DS最常见的原因。只有80%的DS患者在SCN1A外显子区检测到致病性变异,这表明SCN1A内含子区的变异可能导致某些DS患者的疾病发生。针对640名DEE患者的基因组分析发现,在五名患者发现了SCN1A 20号内含子深处高度保守区域内的罕见变异,导致此区域内一段64 bp片段(20N)可以选择性剪接进SCN1A的mRNA里。先前关于DS患者内含子变异和毒外显子的研究是在人工构建的非神经元培养细胞中进行的,不能准确反应生物体内毒外显子产生与DS表型关系,因此本研究通过对导致20N产生的SCN1A内含子变异小鼠模型的构建和分析,为内含子变异与DS发生提供了严格的因果关系证据,直接检测体内毒外显子的转录情况,并为钠离子通道基因毒外显子的功能及其与人类遗传疾病的关系提供新的见解。该项研究2021年1月7日发表在学术期刊PLOS Genetics上,题目为《Aberrant regulation of a poison exon caused by a non-coding variant in a mouse model of Scn1a-associated epileptic encephalopathy》。

研究结果:20N及侧翼区域在进化上高度保守
多物种的序列比对结果表明SCN1A 20N及其侧翼区域高度保守,这提示该区域的变异可能影响Nav1.1的正常生物学功能。当20N及其侧翼序列发生变异时会导致一段64nt的序列被异常剪接进SCN1A 转录本中形成外显子20N,20N位于连接第三个SCN1A同源结构域D3的第四和第五跨膜电压敏感区的胞内环中,20N的滞留引入了一个提前终止密码子,这可能导致无义介导的mRNA降解(图1)。本研究针对Carvil等2018年报道的引起20N产生的变异SCN1A_c.4002+2451G>C,使用CRISPR/Cas9基因编辑技术在小鼠中获得相同位点c.3969+2451G>C的突变,并将携带该变异的小鼠称为SCN1A+/KI以用于后续研究。

图1 20N及其侧翼区域在不同物种间高度保守
20N导致NMD发生并引起小鼠DS
对SCN1A+/KI新出生小鼠脑组织中SCN1A转录水平和蛋白表达水平的检测发现c.3969+2451G>C变异导致小鼠脑组织中SCN1A的转录水平下降为野生型小鼠的50%,针对Nav1.1的蛋白质印迹试验也表明SCN1A+/KI小鼠中全长Nav1.1蛋白(260KD)的积累量下降为野生型小鼠的一半(图2)。有趣的是在SCN1A+/KI小鼠中并未检测到截短蛋白(157KD)的产生,因此c.3969+2451G>C变异导致Nav1.1的降低是由于含有20N的转录本发生无义介导的mRNA降解。
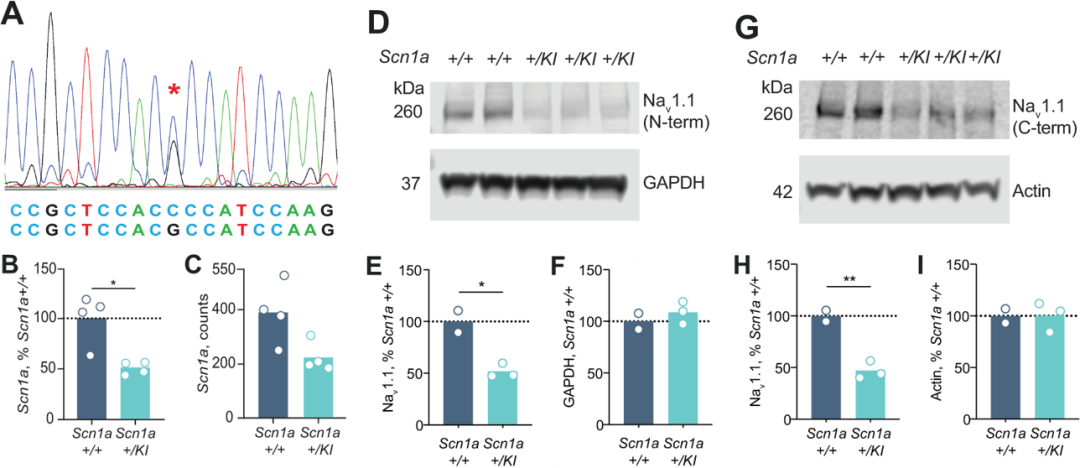
图2 SCN1A+/KI小鼠脑组织中SCN1A 转录本和Nav1.1蛋白水平降低
与野生型小鼠相比,SCN1A+/KI小鼠的预期寿命显著缩短。与其他DS成年小鼠的表型一致,SCN1A+/KI小鼠表现出过度活跃现象,在开放领域的行走距离和垂直跳跃次数均显著高于野生型小鼠。与野生型小鼠相比,SCN1A+/KI小鼠在开阔视野中心的时间百分比和刻板印象计数相似,均没有明显的焦虑增加迹象(图3)。综合小鼠模型的分子生物学检测结果和表型观测结果,证实了毒外显子20N导致小鼠DS的发生。
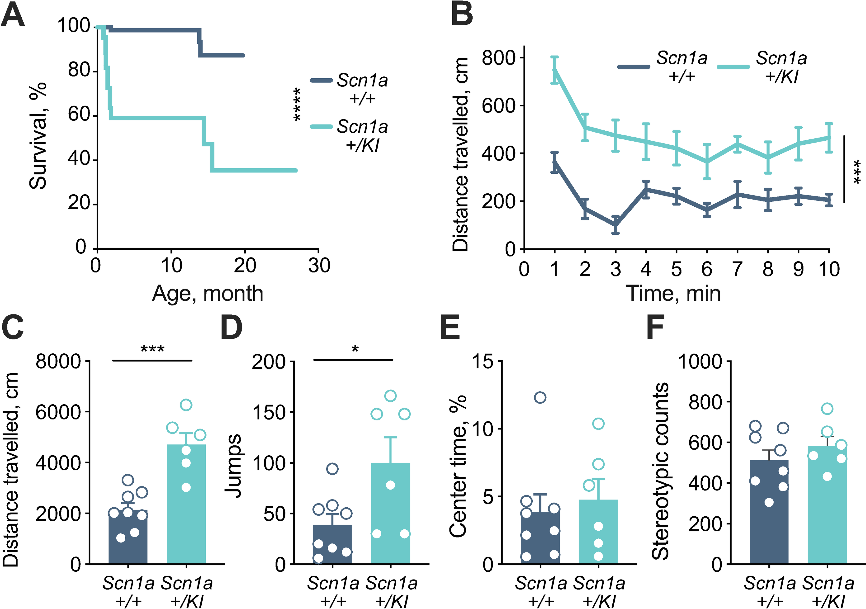
图3 SCN1A+/KI小鼠表现出寿命缩短和多动症表型
20N特异性的在小鼠脑组织中表达并随发育进程逐渐降低
在SCN1A+/KI成年小鼠的脑组织中SCN1A的转录本有4.8%是包含20N的,而在野生型成年小鼠脑组织中这一比例仅为0.97%(图4)。而在脑组织的中,SCN1A+/KI小鼠的SCN1A表达量仅为野生型小鼠的50%,这意味着几乎所有的包含20N的转录本会被降解,不翻译成截短的蛋白。
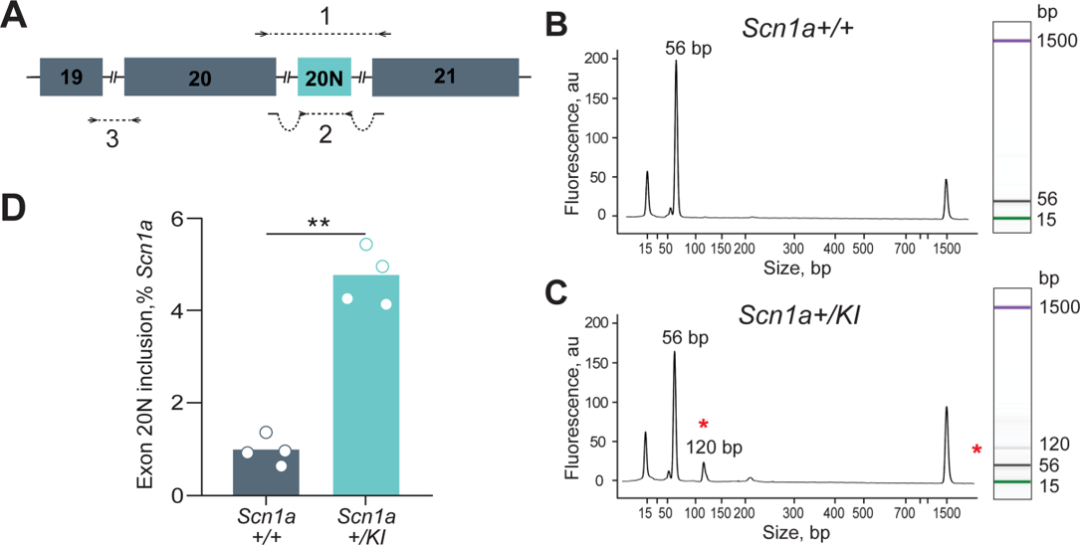
图4 SCN1A+/KI 小鼠脑组织中20N的表达量增加
SCN1A正常转录本在小鼠肺、肝、心脏和肾脏中均有低水平的表达,而在脑组织中表达量最高,20N在野生型小鼠肺、肝、心脏中不表达,在肾脏中仅有非常低水平的表达,在脑组织中表达水平的较高但是低于正常转录本的表达量。而在SCN1A+/KI小鼠的肺、肾脏和心脏中均可检测到20N的表达,在脑组织中的表达水平亦高于野生型小鼠,但在脑组织中SCN1A正常转录本的表达量则低于野生型小鼠。
通过对已公布的非20N变异小鼠皮层多个发育节点的转录组测序数据集来分析小鼠脑组织中20N和正常转录本的表达量,发现小鼠胚胎发育14.5天时,20N在脑组织中表达量最高,包含20N的转录本占SCN1A所有转录本的比例高达70%,随后20N的表达量逐渐降低至出生后30天时20N占比<10%并维持这一水平至成年(图5)。随着异常剪接导致的20N的减少,更多正常剪接形式的SCN1A转录本产生。
这意味着包含毒外显子20N的异常剪接不是偶发事件,而是一种在胚胎早期发育过程中抑制SCN1A表达的主动调控过程。导致毒外显子产生的变异维持了SCN1A的胚胎调控模式,包含20N的转录本持续产生,导致了新生儿SCN1A相关疾病的发生。SCN8A毒外显子18N的表达模式与SCN1A 20N表达模式高度一致,这意味着毒外显子主动调控模式可能是调控神经发育基因精准表达的一个共同特征。
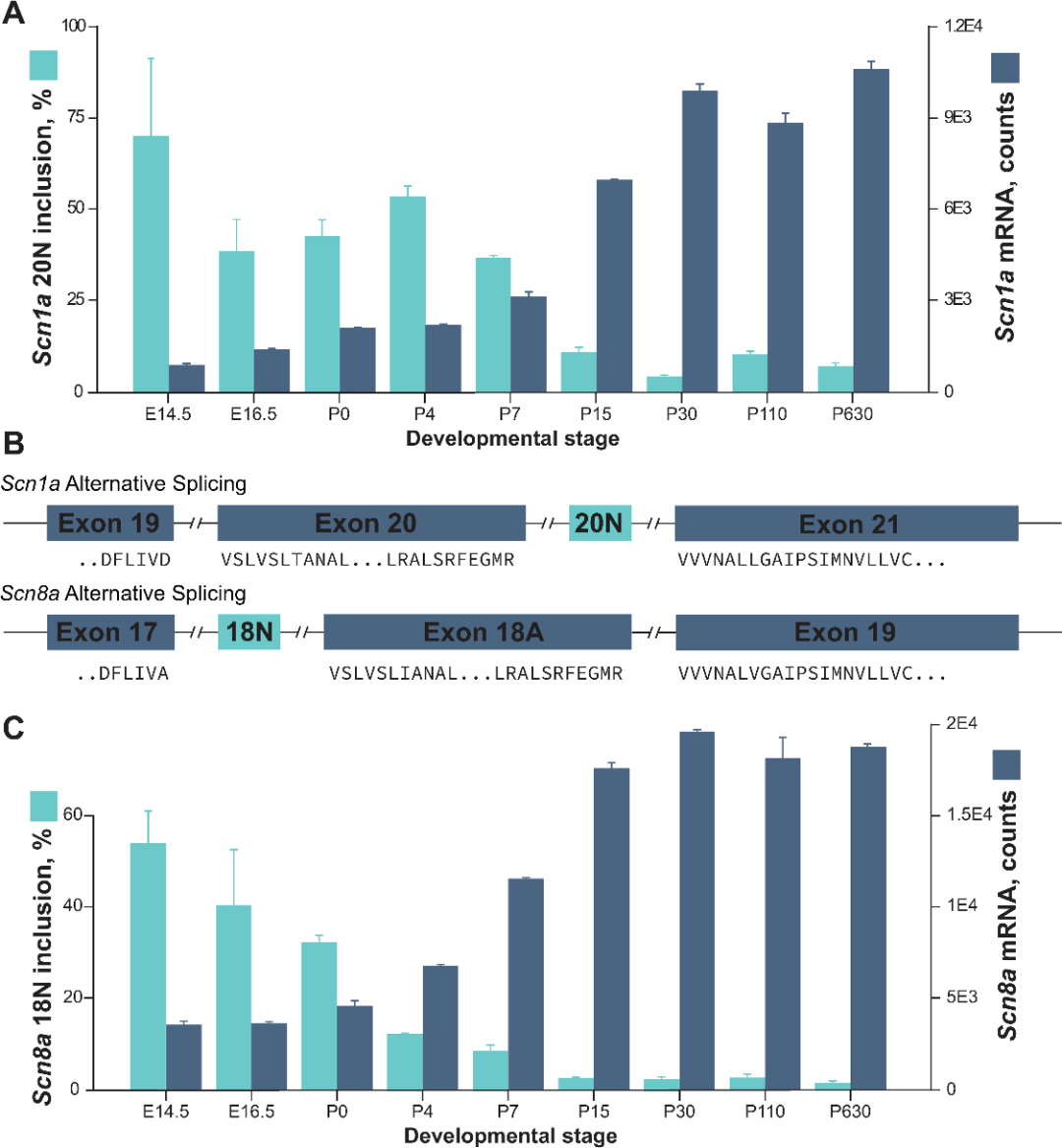
图5 小鼠脑发育过程中钠离子通道基因毒外显子的表达
讨 论
综合本研究与先前关于Nav1.1和Nav1.6的细胞特异性和细胞区域类型特异性的表达研究结果,毒外显子是一种正常且精确的调控模式,可以确保调控神经系统发育基因的时空精准表达,并为SCN1A或SCN8A变异导致临床特征出现的时间提供了一种解释机制。这种机制在进化上是保守的,代表了一种未被认识的孟德尔病的潜在来源。
在胚胎发育早期,多达70%的SCN1A转录本包含毒外显子20N,随着时间的推移这些转录本在出生后减少到10%,这是胚胎早期发育过程中抑制SCN1A表达的主动调控过程。在正常发育过程中,包含20N的转录本在出生后逐渐减少,SCN1A的表达从毒外显子介导的抑制中解除,编码功能蛋白的SCN1A转录本迅速增加。导致毒外显子产生的SCN1A内含子变异不会产生一种全新的正常转录,而是维持了SCN1A的胚胎调控模式,即“持续毒外显子产生”(图6)。这种由于内含子变异引起的持续的毒外显子产生导致了SCN1A正常转录本的表达量下降,具有完整生物学功能Nav1.1合成不足从而导致SCN1A相关疾病的发生。
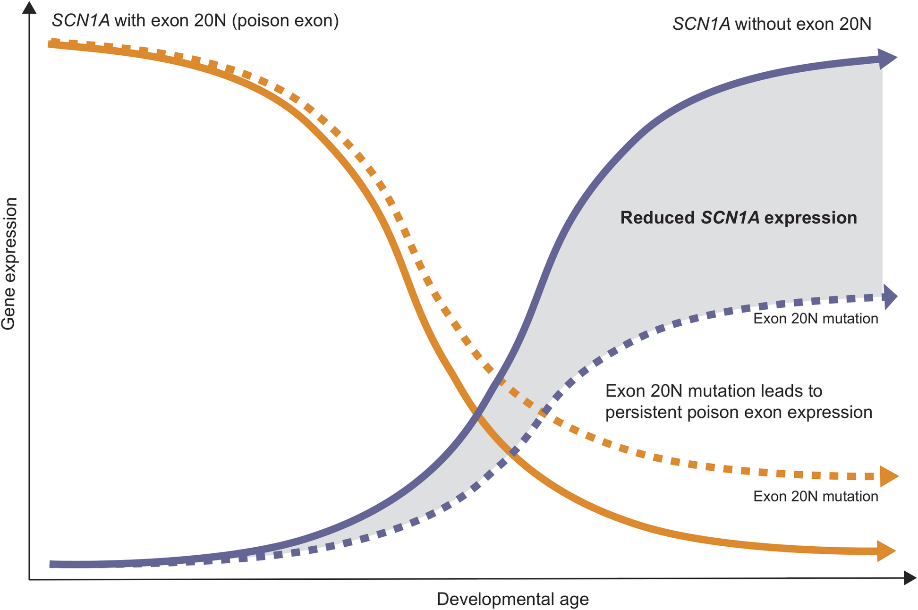
图6 SCN1A 20N转录本表达量随发育进程逐渐降低
单倍剂量不足是DS的疾病机制,两项发现彻底改变了目前对SCN1A相关疾病的认识:在Dravet综合征患者中发现SCN1A高度保守内含子区域的变异导致毒外显子20N的增加; 另一项发现是正常生物体内存在的非功能性的SCN1A 20N异常剪切事件可以通过专门设计的反义寡核苷酸(ASO)减少。这种机制可以在治疗上加以利用,一种针对异常剪接转录本的新的治疗方法即靶向提高该基因表达(Targeted Augmentation of Nuclear Gene Output,TANGO)的方法,在DS小鼠模型中恢复了SCN1A的表达量,减少了癫痫发作和癫痫猝死,这项新技术目前正走向临床试验。
在所有人类基因中,有高达30%的基因转录毒外显子,这似乎是生物体的一种正常调控机制,毒外显子在1200多个与人类疾病相关的基因中也被发现,包括发育性和癫痫性脑病(DEE)的已知基因,如SYNGAP1、SCN2A、SCN8A等。这意味着毒外显子在癫痫基因中很常见,是潜在的治疗靶点。通过动物模型的研究,可以更加深入的探究毒外显子与孟德尔病相关性的,更好地理解疾病的分子发病机制并对疾病治疗靶点的发现和新药物的开发至关重要。
赛福说
遗传病的致病机制复杂多样,越来越多的深度内含子区致病变异被报道。纵观SCN1A毒外显子20N发现过程,基因UTR区域和内含子区域的保守序列在基因的表达和编码蛋白正常生物学功能上起到重要调控作用,目前常规WES测序往往不能覆盖这些区域。因此赛福推出了特别针对SCN1A的毒外显子检测产品,以帮助临床寻找疾病的致病原因。我们已在数例Dravet/FS/GEFS+患者中成功地检测到了引起SCN1A毒外显子产生的深度内含子变异。同时赛福还搭建了转化医学实验室,通过一对一的个性化服务模式,为医生提供基因功能验证的整体解决方案。